

regarding sickle cell disease, treatments, and possible complications
How Sickle Cell Can Affect your Body
Select a part of the body to read more
How Sickle Cell Can Affect your Body | Select a part of the body to read more
Sickle cell anemia affects different people differently and different parts of the body differently. While everyone affected has a different experience, the Body Map aims to show how different parts of the body are most commonly affected by sickle cell anemia.
The Brain | Select a part of the body to read more
The most severe neurologic complication of sickle cell disease is stroke. A stroke can occur if sickled red blood cells “get stuck” in the blood vessels of the brain and block blood flow to the brain. Without treatment, approximately 11% of children with sickle cell anemia (HbSS or HbSβ0-thalassemia) will have a stroke before they are 15 years of age. Thankfully, we now carefully screen for stroke (with transcranial Doppler or TCD studies) and use treatments such as hydroxyurea beginning early in life. These screening tests and treatments have greatly reduced the risk of stroke. Although it is now uncommon in children, it is important to know the signs and symptoms of stroke
If you notice any of the following things, seek care immediately. A stroke is an emergency- Paralysis or weakness on one side of the body or face
- Slurred speech
- Not using an arm or leg (“limping”) but not having pain in that arm or leg
- Loss of consciousness (passing out)
- Severe headaches
- Confusion or not acting right
In addition to stroke, other less obvious damage to the brain can occur as a result of sickle cell disease. Children with sickle cell disease may have what is called a “silent stroke” seen on brains scans (like MRI). This means that a small area of the brain was damaged but did not cause any weakness or paralysis or pain when it happened. Children also may have developmental or academic (school) challenges resulting from sickle cell disease. Early treatment with hydroxyurea is recommended to protect this important brain tissue. We also screen for these other neurologic complications with a brain MRI (starting at age 5 when the child can have an MRI without sedation) and neuropsychological testing to fully assess your child’s learning abilities.
Skin, Hair, and Nails | Select a part of the body to read more
Sickle cell disease does not affect your hair. Hydroxyurea does not significantly affect your hair either. A small proportion of patients may experience mild changes like hair thinning or mild hair loss when they start taking hydroxyurea. These symptoms are usually very mild and should not stop you from taking the medication. If you experience hair changes, talk to your doctor before stopping your medication.
Sickle cell disease does not affect your nails. Hydroxyurea can sometimes make the nails appear dark, but it does not damage the nails, and the color change goes away when you stop hydroxyurea. If you have concerns about nail changes, talk to your doctor before making changes to your medications.
Sometimes sickled cells damage the blood flow to the skin, especially in the legs, and it is difficult for the skin to stay healthy or to heal after an injury. Sometimes people with sickle cell disease develop a skin wound that takes a long time to heal called a skin ulcer (link to vasculopathy). This requires good wound care (bandages, cleaning), pain management, and treatment of the underlying sickle cell disease.
The Lungs | Select a part of the body to read more
Acute Chest Syndrome is a common problem that often begins with or during a pain crisis. It can also happen without pain elsewhere in the body. It is a pneumonia-like illness caused by sickle cells becoming trapped in the blood vessels of the lung. In young children, it is commonly triggered by either viral or bacterial lung infections. Other causes or triggers include anesthesia for surgery, not taking deep breaths (because of pain or sleepiness), being overly sedated or too sleepy (because of pain medications or feeling sick in general), or blood clots or fatty bone marrow particles being trapped by the lungs. Sometimes, acute chest syndrome happens without a known cause or trigger, especially in older children and adults.
Acute chest syndrome causes chest pain, cough, fever, and low oxygen levels. Chest x-rays show changes in the lungs that look a lot like infection. We always treat with antibiotics even though we can’t be sure there is infection deep in the lungs.
Acute chest syndrome can be prevented! When sickle cell patients have pain, they often do not breathe deeply or are sleepy from pain medications. This results in lungs that are not filled with enough oxygen and causes sickling inside the lungs. It is important to use incentive spirometry during pain crises both at home and in the hospital to prevent this complication.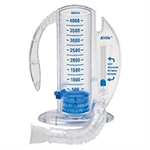
Symptoms:
- Chest pain
- Can’t take deep breaths
- Problems breathing
- Coughing /wheezing
- Fever
If your child has any of these symptoms, it is important to call your sickle cell team or seek care for further evaluation.
Treatment and Prevention:
- Patients diagnosed with acute chest syndrome are usually hospitalized and receive antibiotics and sometimes require oxygen or blood transfusions. Sometimes acute chest syndrome can be serious and require transfer to the intensive care unit (ICU), which is why prevention and early treatment are so important!
- Hydroxyurea reduces the risk of having acute chest syndrome and reduces the risk that acute chest syndrome will result in more serious illness.
- Patients who need anesthesia for surgery or other procedures need special care before and after surgery to prevent acute chest syndrome. It is always necessary to have a specific medical plan for any surgery or procedure which may include a blood transfusion before anesthesia in some patients.
Asthma is not directly related to sickle cell disease, but many children with sickle cell disease also have asthma. Patients with both sickle cell disease and asthma can have a higher risk of lung complications such as acute chest syndrome and even painful episodes (“pain crisis”). If your child has asthma, it is important to see a doctor for it and take any medications as prescribed to protect the lungs and avoid serious complications.
Pulmonary Hypertension is a chronic and serious complication of untreated sickle cell disease. As with all organs of the body, without treatment, sickle cell disease causes slow and progressive damage to the lungs and the blood vessels. This damage, in addition to frequent episodes of acute chest, can damage the blood vessels in the lungs and the lung tissue. Pulmonary hypertension is the most serious of these complications and results in serious lung complications in many adults with sickle cell disease. It is difficult to reverse once it has happened. The early use of treatments for sickle cell disease, like hydroxyurea, with good adherence throughout life can protect the lungs from chronic complications such as pulmonary hypertension.
The heart and lungs work together as a unit, and many times heart disease can cause pulmonary hypertension.
Stomach and Digestive Organs | Select a part of the body to read more
Constipation (firm or hard stools that are hard to pass) is a frequent problem for people with sickle cell disease. Causes of constipation include dehydration and opioid pain medications.
Symptoms
- Passing stool (pooping) less than once a day
- Difficulty passing stools
- Firm, hard or small stools
- Belly pain
Prevention
- Drink plenty of fluids throughout the day
- Drink more when it’s hot or when exercising or playing
- Carry a water bottle
- Eat foods that are rich in fibers
Treatment
- Laxative medications such as MiraLax or senna as needed
- Some people may need to take laxatives on a schedule to stay regular
Abdominal or belly pain can have many different causes related to sickle cell disease, including constipation , sickle cell pain (“crisis”), gallstones, kidney problems, or spleen problems. It can also be related factors not caused by sickle cell disease such as heartburn. The most important thing to do is figure out what the cause is and treat or manage that specific cause. Sometimes figuring out the cause of the abdominal pain can take some time.
Kidneys & Bladder | Select a part of the body to read more
The kidneys allow the body hold on to fluid (water) and get rid of waste in the urine. Damage to the kidneys starts early in life. People with sickle cell disease can’t concentrate their urine (hold on to enough water), so they urinate more than other people. This can lead to dehydration. So, people with sickle cell disease need to drink more water to stay hydrated and healthy. The extra urination means more trips to the bathroom (schools and employers need to accommodate this) and sometimes bedwetting (“enuresis”) will continue beyond early childhood.
Symptoms of Dehydration:
- Dry, sticky mouth and lips
- Tiredness
- Urinating much less (e.g., fewer wet diapers)
Prevention
- Drink plenty of fluids throughout the day
- Drink more when it’s hot or when exercising or playing
- Avoid drinks with caffeine
- Carry a water bottle
Treatment
- Give your child extra fluids to drink, especially water
If you are dehydrated, your urine will be more concentrated and darker. This is a reason to drink more fluids. Urine may also be darker or redder than people without sickle cell disease, because the breakdown products of hemoglobin (from the anemia of sickle cell disease) can be passed in the urine. Sometimes this change in color is more prominent during a painful episode. This is not a problem. The main thing to rule-out is blood in the urine.
Sometimes even tiny areas of kidney damage can cause blood to appear in the urine. This blood can be invisible to you and only seen when a lab test is done on the urine. Sometimes there is enough blood to be visible. Occasionally, there may be a large amount of blood that may last for days to weeks. Blood in the urine is usually painless, but sometimes back, flank, or abdominal pain can happen. Seek medical attention if you think you have blood in your urine.
Sickle cell disease can also damage the kidneys so they can’t do the job of getting rid of bodily waste. The kidneys also hold onto important things such as protein. One of the earliest signs of kidney disease is the leakage of protein (such as albumin) into the urine. Urine should be checked for protein regularly during clinic visits (1-2 times per year). If kidney disease is untreated, it can lead to kidney failure and even the need for dialysis.
Symptoms of kidney failure
- Fatigue
- More anemia than usual
- Increase or decrease in amount of urine
Prevention of kidney failure
- Good, ongoing treatment of sickle cell disease (hydroxyurea, chronic transfusions)
- Screening during routine visits before severe damage occurs
- Oral medicines (ACE inhibitors and ARBs) for people with a lot of protein in the urine
Jaundice, Liver and Gall Bladder | Select a part of the body to read more
People with sickle cell disease often have jaundice, which is most easily seen as a yellow discoloration of the eyes. The yellow color itself is harmless, but it is a clue about something happening in the body. This yellow color comes from a substance that we all have in our body called bilirubin. Bilirubin is removed from the body by the liver. High levels of bilirubin can result from two main problems: red blood cell problems (like sickle cell disease) or liver and gallbladder problems. When red blood cells are broken down or destroyed quickly, as they are in sickle cell disease, bilirubin is made faster than the liver can get rid of it. When that happens, the person becomes jaundiced. If a person with sickle cell disease is sick or has a pain crisis, they may have more jaundice around that time.
Gallstones are a complication of sickle cell disease. When red blood cells (sickle cells) are destroyed, bilirubin is made. This causes jaundice (the yellow color). All the extra bilirubin goes to the liver to be removed from the body. The liver gets rid of it through the bile ducts and the gallbladder. Making a lot of bilirubin all the time can cause stones made out of bilirubin to be formed in the gallbladder and bile ducts.
Symptoms:- Sudden, severe pain in the upper right or center of your abdomen, just below your breastbone and ribs, especially after eating
- Back pain between your shoulder blades, especially after eating
- Pain in your right shoulder, especially after eating
- Nausea or vomiting, especially after eating
- Eat a healthy diet
- Drink plenty of fluids
- Keeping your sickle cell disease well-controlled with treatments like hydroxyurea
- Removing the gallbladder (surgery)
- Sometimes medications can be tried before surgery
Sickle cell disease can cause several problems in the liver, such as scarring and inflammation. Sometimes this is a chronic or long-term problem. Sometimes it can be an acute (sudden) problem. Problems with the liver can be detected by laboratory tests. A sudden increase in the amount of jaundice and enlargement and tenderness of the liver (on the right side of the abdomen at and below the ribs) can mean a liver problem. Imaging (pictures) of the liver may be needed, by ultrasound, for example. Treatment depends on the type of liver disease. Like most problems in sickle cell disease, we think that keeping your sickle cell disease well-controlled all the time can prevent many liver problems.
Some people with sickle cell disease have iron overload in their liver from many blood transfusions. If that happens, it is important to take medicine (iron chelation therapy) to remove excess iron from the liver and prevent liver damage. Liver iron content can be monitored with yearly MRIs as well as lab values.
Bones | Select a part of the body to read more
Blood flows through the center of your bones (the bone marrow) to keep your bones healthy and strong. When blood flow is blocked inside the bones you feel pain. Even when the blockages are small, you can have severe pain. Over time, the blockages damage the blood vessels and blood cannot flow to all areas of the bone. The bone cannot stay healthy without the blood flow. The bone becomes unhealthy, weak, and may break more easily. This is called avascular necrosis. The large bones in your hip joint and shoulder joint need a lot of blood flow to keep them healthy, and they are the most likely to develop avascular necrosis, but any bone can be affected.
Symptoms of avascular necrosis include
- Pain around the joints (hips, knees shoulders)
- Swelling sometimes is seen around the knee joint
- Limping
- Difficulty using the affected joint with limited movement
The bones can also become infected when bacteria get stuck inside the abnormal blood vessels inside the bone marrow. A bone infection is called osteomyelitis. When you have osteomyelitis, you need to use antibiotics for a long time (several weeks) to get rid of the infection.
Symptoms of osteomyelitis
- Pain in the affected bone
- Swelling
- Fever
- Inability to use the affected limb
Reproductive Organs | Select a part of the body to read more
Sickled cells can cause a blockage in the small blood vessels that keep your reproductive organs healthy. In both boys/men and girls/women, the pituitary gland in your brain is the main control switch that sends signals to your reproductive organs. It tells your body when to start puberty and sexual development, and it helps control the reproductive cycle in girls/women. In the boys/men, the testes produce sperm. If the testes are damaged, the number of sperm released during ejaculation can sometimes be lower. In girls/women, the ovaries produce eggs. If the ovaries are damaged, the number of eggs available can be lower so that they stop releasing eggs sooner than other healthy women. Also, the timing of the release of eggs can be changed so that it is more difficult to get pregnant. Most people with sickle cell disease do not have any problems having children, but if you do have problems, a specialized doctor can help. Hydroxyurea can increase the number of healthy blood cells and decrease the amount of damage to reproductive organs. People who are treated with a bone marrow transplant must take strong medicine before the transplant that can damage their ovaries or testes. This must be discussed with your sickle cell doctor and the transplant doctor before you start.
Menstrual symptoms can provoke sickle cell pain in some people. A gynecologist is an important part of the multidisciplinary team for young women with sickle cell disease, beginning in the teenage years. A gynecologist can help develop a plan for menstrual suppression to prevent increased sickle cell pain.
In males with sickle cell disease, sickled cells can cause blockages in the blood vessels of the penis, causing a long lasting, painful erection, called priapism. This is not related to sexual arousal and can occur even in younger boys prior to puberty.
Symptoms
- An erection lasting longer than expected
- A painful erection not brought on by typical sexual arousal
Treatment at Home
- Drink fluids
- Pass Urine
- Take a warm bath or shower
- Walk around
- Avoid alcohol / cannabis / tobacco / Viagra®, as these can potentially worsen or trigger priapism
Prevention and Further Management:
There are a number of common medicines that treat or prevent priapism. Your hematologist or urologist will talk to you about what is the best treatment to manage your type of priapism. If the episode of priapism lasts longer than 2 hours seek medical attention immediately.
Heart | Select a part of the body to read more
Without treatment, sickle cell disease results in slow and progressive damage to all organs, including the heart. For the heart, this damage is very slow and usually not noticed until later in life, often not until the teenage or adult years. However, when we look at the hearts of sickle cell patients with special tests, we notice that this damage begins early in life. We don’t yet know how to reverse the damage once it has occurred, but we think it can be prevented in many cases by early treatment of sickle cell disease. It is important to start treatment with sickle cell medications, such as hydroxyurea, early in life and continue these treatments throughout life to keep your heart healthy. We screen for heart problems with an echocardiogram (“echo”), electrocardiogram (ECG or EKG), and sometimes a heart MRI at approximately 18 years of age.
Heart problems from sickle cell disease include enlargement of the heart, fibrosis or scarring of the heart muscles, conduction (“electrical”) abnormalities causing abnormal heart rhythms, pulmonary hypertension and, when sickle cell disease is poorly controlled, heart failure and sudden death.
The Eyes | Select a part of the body to read more
It is common for people with sickle cell disease, especially those who do not receive treatments, to have a yellow color of the usually white part of the eyes. The medical term for this is “scleral icterus”. When red blood cells break apart, which happens in sickle cell disease, a substance called bilirubin is made. Bilirubin is yellow and causes jaundice. This is not dangerous and does not cause any problems by itself, but is a sign that there are a lot of sickled red blood cells in your body. Treatment with medications like hydroxyurea reduces the yellowing of the eyes and jaundice.
The back of your eye has a very thin layer that senses light and helps you see. This is called the retina. Very small blood vessels in the back of your eye carry blood to the retina and keep this layer healthy. If sickle cell disease is not well-treated, sickled red blood cells can get trapped in these blood vessels. The blood vessels are damaged, blocking blood flow, so oxygen cannot get to the retina to keep it healthy. When the retina is damaged over time, it causes vision problems (sickle retinopathy) which can even lead to blindness if not treated. Your body tries to make new blood vessels to keep the retina healthy, but the new blood vessels are weak and can bleed into the eye easily. Symptoms of retinal damage typically begin in the teenage and adult years and are common for all types of sickle cell disease. Vision problems are among the most common problems for people with HbSC disease.
- Symptoms: often there are no symptoms in early retinopathy. When they do develop patients report:
- Blind spots
- Blurred Vision
- Increase in “floaters”
Prevention: We recommend yearly eye exams by an eye doctor (ophthalmologist) beginning at 8-10 years of age. If the eye doctor finds any damage, they will tell you how often they need to look at the eyes and whether eye treatment is necessary.
Treatment: Early treatment of sickle cell disease with hydroxyurea is important to prevent damage to all parts of the body, including the eye. Your eye doctor may also use laser therapy to treat abnormal blood vessels.
Blood Vessels | Select a part of the body to read more
Anemia is a term that describes fewer red blood cells or decreased hemoglobin. The abnormally shaped red cells in sickle cell disease are easily broken by the body and live shorter than normal red blood cells. This causes a decrease in the number of red cells (anemia). All patients with sickle cell disease have anemia, but the severity of anemia varies from one individual to another. When anemia is severe, the patient may experience fatigue, poor energy or decreased oxygen levels. The severity of anemia in sickle cell disease is an indicator of the severity of sickle cell disease in general, and can increase the risk of organ damage in some individuals.
Some individuals may have acute or rapid worsening of their anemia that requires a blood transfusion. The most common example is when an individual has a viral infection (e.g., parvovirus B19) which slows down red blood cell production for a period of time and causes severe anemia. This condition is called acute aplastic crisis. Patients who experience this complication usually require blood transfusion (sometimes more than once) until their body gets rid of the virus and they start producing red blood cells at their normal rate.
Acute splenic sequestration is another cause of acute anemia due to blood trapping in the spleen and sudden enlargement of the spleen. This is often treated with blood transfusions.
The sickle cell disease medications hydroxyurea and Voxelotor (Oxbryta®) both increase the hemoglobin and improve anemia symptoms in individuals with sickle cell disease.
White blood cells and platelets are other types of blood cells the body produces in addition to red blood cells. The main function of the white blood cells is to fight infections while platelets prevent or stop bleeding. Both white blood cells and platelets increase when there is inflammation in the body and are often increased in individuals with untreated sickle cell disease. This increase is more notable during a pain crisis. Among the many benefits of hydroxyurea in sickle cell disease is a reduction or normalization of white blood cells and platelets.
Vasculopathy sickle cell disease affects all organs in the body including the blood vessels. Vasculopathy is a term that means damaged blood vessels which can occur in individuals with sickle cell disease due to repeated sickling and red cell breakdown. The damaged blood vessels in the brain increase the chance of developing strokes. Other complications that are linked to vasculopathy or damaged blood vessels in individuals with sickle cell disease include pulmonary hypertension, priapism and leg ulcers.
Ear, Nose and Throat | Select a part of the body to read more
Obstructive sleep apnea is a common sleeping disorder that happens when the throat muscles relax too much and block the airways during sleep. This causes intermittent pauses in breathing (apnea) while sleeping. Obstructive sleep apnea is not directly caused by sickle cell disease, but many individuals with sickle cell disease also have obstructive sleep apnea. Snoring is one of the common symptoms of sleep apnea, but not everyone who snores has obstructive sleep apnea.
Symptoms of obstructive sleep apnea include
- Loud snoring
- Stopped breathing during sleep
- Gasping or choking at night
- Excessive daytime sleepiness
- Difficulty concentrating during the day
- Morning headaches
- Nocturnal enuresis
Many patients with obstructive sleep apnea do not know that they have it. Your doctors will screen for obstructive sleep apnea by asking about these symptoms during clinic visits. In addition to these symptoms, some patients may be suspected to have obstructive sleep apnea if they have unexplained low oxygen levels during sleep or when lying down. This is often picked up when patients are hospitalized for other reasons and have their oxygen levels monitored while sleeping. If sleep apnea is suspected, you may be referred to a sleep specialist who will evaluate the symptoms and may perform an overnight sleep study.
Treatment
Untreated sleep apnea negatively affects the quality of life, reduces productivity, and increases the risk of cardiac disease and high blood pressure. Treatment of obstructive sleep apnea is variable from one patient to another and may include tonsillectomy in young children, weight reduction in overweight individuals, oxygen supplementation, and using a device that applies pressure to keep the airways open during sleep (called positive pressure ventilation). Hydroxyurea reduces low oxygen (hypoxia) in some patients and may help these symptoms.
Hearing is not affected in people with sickle cell disease. However, some patients who receive certain medications, especially the medications that treat iron overload (hyperlink to iron overload section), can have hearing problems. If you receive one of these medications (e.g., Desferal® or Jadenu®) you will be getting hearing tests regularly to screen for early signs of hearing problems.
Milestones | Select an icon to read more about milestones and personal experiences
Milestones
Select an icon to read more about milestones and personal experiences
.png)
.png)
.png)
.png)
.png)
.png)
.png)
.png)
.png)
.png)
.png)
.png)

Every baby born in the USA is screened for sickle cell disease at birth using a test called hemoglobin electrophoresis. The test is done at least once at the time of birth (usually on the first or second day of life when your baby is still in the hospital). If the test is abnormal in any way, it will be repeated before 2 months of age to make a diagnosis. If needed, genetic testing is also done to be sure about the diagnosis.
FAQs
Sickle cell disease damages the spleen very early in life. The spleen is part of the immune system that helps fight bacteria (infection) in the bloodstream. Young babies and children with sickle cell disease are more likely to get sick from these types of infections. To prevent these infections, you child will take penicillin by mouth twice a day until age 5 (and get other helpful and safe vaccines for more protection).
FAQs
Routine immunizations at the pediatrician are very important to prevent infections and viruses that can be even more serious in a person with sickle cell disease. At the sickle cell clinic there are a few extra vaccines recommended to prevent against pneumococcal pneumonia and meningitis (bad infections of the lungs and brain). Both penicillin and immunizations are important to reduce infection risk in people with sickle cell disease. Even if an individual is immunized and taking penicillin, they should be evaluated immediately if they develop fever ≥101.5 F to rule out blood stream infection (bacteria in the blood).
FAQs
Hydroxyurea is life saving and important medication for those living with sickle cell disease. It is recommended for all children with severe sickle cell disease starting at 6 months of age. Hydroxyurea can also be beneficial for those who have milder forms of sickle cell disease, too.
Hydroxyurea works by increasing fetal hemoglobin in the body. This results in improved blood counts and less anemia. It also reduces “sickling” and pain. Hydroxyurea aids in the reduction and prevention of both short and long term complications of sickle cell disease.
Those who take hydroxyurea have less pain and visits to the hospital. Overall they are healthier and have an improved quality of life. Hydroxyurea is taken one time every day and is available as a liquid, tablet, or capsule.
FAQs
TCD studies are non-invasive ultrasound tests (images) looking at blood-flow in the brain. The results of TCD helps the medical team estimate the risk of developing stroke. Typically this exam is started around 2 years old and repeated at least yearly during childhood and adolescence. If the results are not normal, your team may recommend repeating TCD sooner. Patients with abnormal TCD results have the highest risk of developing stroke. These results help you and the medical team decide on the appropriate treatment plan.
FAQs
It is important to share with your daycare provider that your child has sickle cell disease. Instruct them to call you or the clinic for any fever >101.5, if the child has pain, swelling in their hands and feet (dactylitis) or seems tired and acting differently than usual. Your clinic may have a team member dedicated to teaching schools about sickle cell disease. Many places have a school intervention specialist, and his/her main job is to explain to schools and daycares they can keep your child healthy and safe.
Some items specific to your child that you can consider sharing:
- if child needs more frequent diaper changes or restroom access
- child displays discomfort/pain (cry, shut down, does not want to be touched)
- when to give medications like ibuprofen
FAQs
An MRI is a picture of the brain, it shows the structure of the brain tissue and the blood vessels. This is an important test because it can show potential complications of sickle cell disease such as strokes or silent strokes that could change the treatment plan. This is a complementary test to TCD (link?) and does not replace it. Generally, a child’s first MRI is around age 5 and repeated every 5 years if the initial MRI is normal.
Neuropsychological testing also begins around 5 years old. This is a series of tests performed by a psychologist that will assess a child's development and learning needs. Some individuals with sickle cell disease may have developmental or learning concerns despite normal brain MRI and normal TCD. After neuropsychological testing is completed, a report will be generated on how to best help your child succeed. It can also help identify their individual strengths and weaknesses. This report can be shared with the school.
FAQs
Children with sickle cell disease should start school at the same time as other children. Your sickle cell team can inform the school about your child’s diagnosis and provide instructions for how to keep you child healthy and safe during school. Your child should stay hydrated, avoid temperature extremes, and rest whenever needed. If your child is having any challenges in school, the sickle cell team can help you find resources and ensure that the school evaluates for any disabilities. The school can develop an individualized education plan (IEP), or a 504 plan so that all the teachers know the best way to help your child.
FAQs
A 504 accommodation plan is a document created collaboratively by the school team and family to outline specific accommodations that will be available to ensure a student with a medical disability has access to the educational settings and curriculum so they can fully participate in the entire school day and all school related activities. It does not include modifications to the way the child learns or displays content knowledge. It does not include specially designed instruction (special education). 504 plans can include special transportation and planning for absences. To pursue a 504, the family should meet with the school team to discuss needs and then follow up with an email to the school team to establish a written record of the request. The school may need to complete an evaluation and meet with the family to determine eligibility for a 504 plan.
An Individualized Education Plan (IEP) is a document created collaboratively by the school team and family that outlines the specific educational needs and accommodations for a student with an educational disability. It is developed collaboratively by the school team and family. An IEP can include all the accommodations on a 504, but additionally includes specially designed instruction (special education). It can also include related services, such as speech therapy, occupational therapy (OT), and physical therapy (PT). To pursue an IEP, the family should meet with the school team to discuss needs and then follow up with an email to the school team to establish a written record of the request. If school agrees, the process includes an evaluation and multiple meetings to determine eligibility for an IEP.
Can include, but are not limited to the following: familiar with pain plan, avoiding triggers (such as extreme temperatures and seating near window or AC vent), no ice for common first aid, monitor for over-fatigue, access to breaks, access to nurse, access to fluids, access to rest room, fever precautions, know symptoms and response steps for splenic sequestration, ACS, and stroke. Depending on how your diagnosis impacts schooling, accommodations could include: modified dress code to allow extra layers of clothing, plan for fire drills to minimize weather exposure, curb-to-curb transportation, elevator access, easily accessible locker, extra set of books for home, and field trip planning. To accommodate absences, consider: extended time, prioritized assignments, and intermittent homebound instruction.
Talk to your provider if you feel your child’s school staff need education on your child’s diagnosis. Educational materials and staff are available to help you educate your child’s school team to increase awareness and understanding.
When you grow and develop your sickle cell symptoms may change. It can be hard to remember to take your medications, but it is very important that you take them every day.
Girls will start having a monthly menstrual period as they go through puberty. During the monthly period, pain can worsen in some patients. If this happens talk with your doctor about ways to decrease pain during a period.
Boys will start having an erection. This is normal. However, sometimes boys with sickle cell disease can have an abnormal erection that lasts too long and is painful called priapism (link?). If this happens talk with your doctor about ways to prevent it.
Women with sickle cell disease can become pregnant and deliver a healthy baby. Sickle cell disease can cause problems during pregnancy, so you will need to see a blood doctor and an obstetrician who are familiar with sickle cell disease during pregnancy. They will help create a treatment plan to keep you and your baby healthy during the pregnancy. They will also help create a safe plan for delivery.
FAQs
It is important to keep your sickle cell disease managed/controlled during pregnancy. Talk to your hematologist if you plan to become pregnant or soon as you become pregnant to create a treatment plan for you during pregnancy.
Yes. Women with sickle cell disease can give birth vaginally. The birth method will be determined by other factors related to the mother and fetus’s health rather than by sickle cell disease only. This will be determined in discussion with the obstetrician at time of delivery.
You do not need to have a c-section only because you have sickle cell disease. The birth method will be determined by other factors related to the mother and fetus’s health rather than by sickle cell disease only. This will be determined in discussion with the obstetrician at time of delivery.
Babies of mothers with sickle cell disease may not have sickle cell disease unless their father’s have sickle cell disease or trait (or other hemoglobinopathy trait), too. If the father does not have sickle any hemoglobinopathy trait or sickle cell disease, children of mothers with sickle cell disease will not have sickle cell disease.
All babies born in USA are screened for sickle cell disease at birth. If the screen is abnormal additional confirmatory testing will be done before the age of 2 months.
People with sickle cell disease can play sports. Some precautions should be taken to decrease the chance of getting sick while you play. Ideally, you should be treated with a disease-modifying medication like hydroxyurea so that your blood and organs are healthy. You should stay hydrated by drinking plenty of fluids and avoid playing too long in hot weather. You should avoid temperature extremes (hot or cold) so that you don’t get pain. It can be helpful to tell your coach that you have sickle cell disease so that they know what to do if you are injured and so that they adjust your training accordingly.
FAQs
As you grow up you will need to find a doctor that has more experience treating adults with sickle cell disease. The medical system for adults (adult emergency departments, hospitals, and clinics) is different than the medical system that cares for children. Your pediatric doctor can help you get ready to move to an adult doctor. The more that you know about your disease and about your treatment, the easier it will be to go to adult care. Preparing for transitioning to adult care is a process that starts in the early teen years.
FAQs
With good planning, people with sickle cell disease can travel to many places. Some patients will travel for their job, some for vacation, or some to study. Some places have fewer resources, but if possible, it is helpful to find a local pharmacy, emergency department, and hospital at your destination. It is important to pack enough of your daily medications to last your whole trip, and you might need extra pain medications. If you use liquid medication, you might need to obtain a refill from a local pharmacy. If you plan to travel, inform your treatment team beforehand so that they can help you prepare for your trip. Your team may write a medical letter that explains your disease and medical needs so that you can show the letter to other medical facilities/ER during travel, if needed.
Resource Toolkit | Select a topic to read more
Resource Toolkit
Select a topic to read more
Outside Resources | Select a site to visit
Outside Resources
Select a site to visit







